User Guide
Importing Alphafold Data
Supported formats
The af_analysis package supports the following file formats for input data:
Importing procedure
Create the Data object, giving the path of the directory containing the results of the AlphaFold results. af_analysis
supports reading the following directory formats:
Alphafold 2 [1]
Alphafold 3 [2]
ColabFold [3]
AlphaPulldown [4]
Chai-1 [5]
Boltz1 [6]
MassiveFold [12]
import af_analysis
my_data = af_analysis.Data('MY_AF_RESULTS_DIR')
For AlphaPulldown and MassiveFold, you should explicitly define the format when importing the data:
import af_analysis
my_data = af_analysis.Data('MY_AF_RESULTS_DIR', format="alphapulldown")
# or
my_data = af_analysis.Data("MY_AF_RESULTS_DIR", format="massivefold")
Alternatively, you can use a python dictionary containing the pdb file list, the .json / .npz data file and the query name.
import af_analysis
pdb_list = [
"test/model_5_seed_002.pdb",
"test/model_5_seed_007.pdb",
"test/model_5_seed_003.pdb",
"test/model_5_seed_006.pdb",
"test/model_5_seed_004.pdb",
]
json_list = [
"test/model_5_seed_002.json",
"test/model_5_seed_007.json",
"test/model_5_seed_003.json",
"test/model_5_seed_006.json",
"test/model_5_seed_004.json",
]
data_dict = {
"data_file": json_list,
"pdb": pdb_list,
"query": "test_amyloid",
}
my_data = af_analysis.Data(data_dict=data_dict)
# Extract the data that you know are present in the json/pickle files
my_data.extract_fields(["plddt", "ptm", "iptm"])
Extracted data are available in the df attribute of the Data object as a pandas dataframe.
my_data.df
Merge Data objects
You can merge multiple Data objects into a single object using the concat_data() function.
my_data1 = af_analysis.Data('MY_AF_RESULTS_DIR1')
my_data2 = af_analysis.Data('MY_AF_RESULTS_DIR2')
my_data = af_analysis.concat_data([my_data1, my_data2])
Scores
pDockq
pDockQ (Predicted DockQ) [7] is a metric that predicts the quality of protein-protein interactions in dimer models. It uses a sigmoid function that considers the number of contacts in the interface (\(log(number \: of \: interface \: contacts)\)) and the average pLDDT (predicted lDDT score) of the interface residues (\(\overline{plDDT_{interface}}\)).
Implementation was inspired from https://gitlab.com/ElofssonLab/FoldDock/-/blob/main/src/pdockq.py.
The pdockq() function calculates the pDockq [7] score for each model in the dataframe.
The pDockq score ranges from 0 to 1, with higher scores indicating better model quality.
where:
\(L = 0.724\) is the maximum value of the sigmoid, \(k = 0.052\) is the slope of the sigmoid, \(x_{0} = 152.611\) is the midpoint of the sigmoid, and \(b = 0.018\) is the y-intercept of the sigmoid.
from af_analysis import analysis
analysis.pdockq(my_data)
For the multiple pDockQ or mpDockQ [8] this values are used: \(L = 0.728\), \(x0 = 309.375\), \(k = 0.098\) and \(b = 0.262\).
from af_analysis import analysis
analysis.mpdockq(my_data)
pDockq2
pDockQ2, or Predicted DockQ version 2 [9], is a metric used to estimate the quality of individual interfaces in multimeric protein complex models. Unlike the original pDockQ, pDockQ2 incorporates AlphaFold-Multimer’s Predicted Aligned Error (PAE) in its calculation, making it more sensitive to large, incorrect interfaces that might have high confidence scores based solely on interface size and pLDDT. pDockQ2 scores range from 0 to 1, with higher scores indicating better interface quality.
with
\(L = 1.31\) is the maximum value of the sigmoid, \(k = 0.075\) is the slope of the sigmoid, \(x_{0} = 84.733\) is the midpoint of the sigmoid, and \(b = 0.005\) is the y-intercept of the sigmoid.
Implementation was inspired from https://gitlab.com/ElofssonLab/afm-benchmark/-/blob/main/src/pdockq2.py.
from af_analysis import analysis
analysis.pdockq2(my_data)
LIS Score
The Local Interaction Score (LIS) [10] is a metric specifically designed to predict the likelihood of direct protein-protein interactions (PPIs) using output data from AlphaFold-Multimer [11]. Unlike metrics like interface pTM (ipTM), which measures the overall structural accuracy of a predicted complex, LIS focuses on areas within the predicted interface that have low Predicted Aligned Error (PAE) values. These low PAE values, often visualized as blue regions in AlphaFold output maps, represent areas of high confidence in the interaction prediction
Here’s how LIS is calculated:
- Local Interaction Areas (LIAs) are identified: Regions of the predicted interface with PAE values below a defined
cutoff (typically 12 Å) are designated as LIAs.
- PAE values within LIAs are inverted and averaged: PAE values within LIAs are transformed to a 0-1 scale, with
higher numbers indicating stronger interaction likelihood. These values are then averaged across the interface to produce the LIS score.
- The LIS method is particularly adept at detecting PPIs characterized by localized and flexible interactions, which
may be missed by ipTM-based evaluations. This is particularly relevant for interactions involving intrinsically disordered regions (IDRs), which are often missed by structure-based metrics.
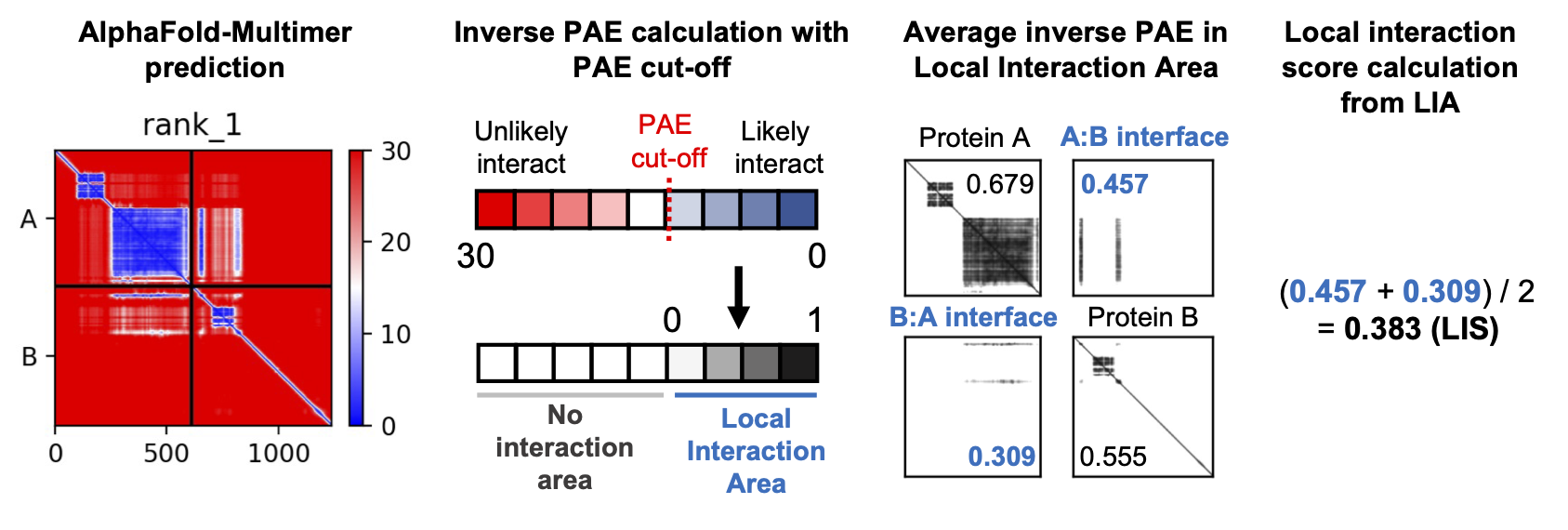
Figure from github.com/flyark/AFM-LIS. Implementation was inspired from https://github.com/flyark/AFM-LIS.
to compute the LIS matrix among subunits:
from af_analysis import analysis
import seaborn as sns
from cmcrameri import cm
# Extract LIS heatmap among subunits
analysis.LIS_matrix(my_data, pae_cutoff=12.0)
# Plot the heatmap
ax = sns.heatmap(my_data.df.LIS.iloc[0], cmap=cm.roma)
ax.collections[0].set_clim(0,1) # Set the heatmap range
ax.set_title('LIS heatmap among subunits')
ax.set_xlabel('Subunit index')
ax.set_ylabel('Subunit index')
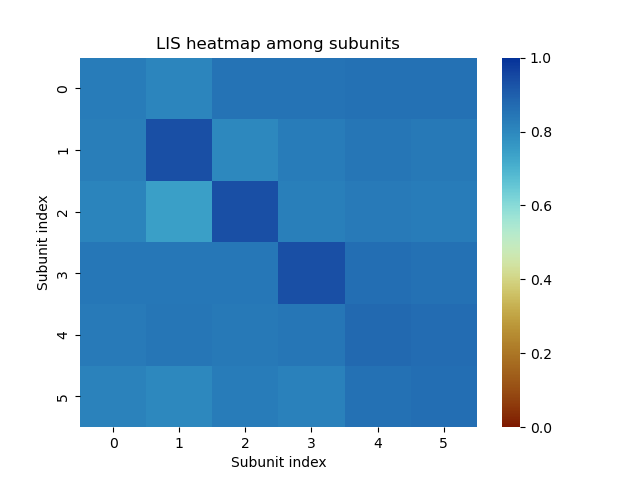
Example of LIS heatmap among subunits on a protein-DNA-Zn complex computed with AlphaFold 3.
actifpTM and chain ipTM
The actual interface pTM (actifpTM), has been included in colabfold version 1.5.5 (march 2025).
If you have used the --calc-extra-ptm option to launch colabfold_batch command,
the actifpTM and chain ipTM scores are available in the colabfold .json output files.
An extra step is necessary to extract the scores from the .json files:
my_data = af_analysis.Data('MY_AF_RESULTS_DIR')
my_data.extract_data()
You should now have access to additional columns in the dataframe:
my_data.df[['pairwise_actifptm', 'pairwise_iptm', 'per_chain_ptm', 'actifptm']]
The
pairwise_actifptmcolumn contains the actifpTM scores for each pair of chains in the model.The
pairwise_iptmcolumn contains the ipTM scores for each pair of chains in the model.The
per_chain_ptmcolumn contains the pTM scores for each chain in the model.The
actifptmcolumn contains the actifpTM scores for each model in the dataframe.
Protein-Protein and Protein-Peptide Docking
The af_analysis package provides a simple interface to score protein-protein and protein-peptide docking
using the docking package.
Note
The docking package infer that the peptide chain or the protein ligand chain is the last one in the model.
The docking package allow to compute:
pae_pep(): average interface of Predicted Aligned Error (PAE) between the receptor chain(s) and theligand/peptide chain (last one). Add the columns
PAE_pep_redandPAE_rec_pepin the dataframe.
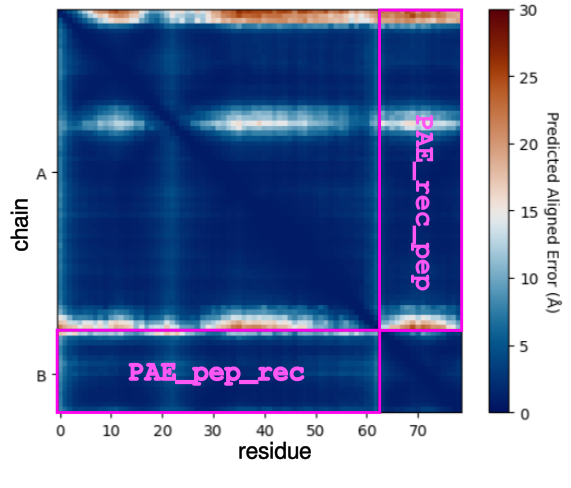
plddt_pep(): compute the average pLDDT of the ligand chain. Add the columnplddt_pepin the dataframe.pdockq2_lig(): compute the pDockQ2 scores of each chain. Add the columnspdockq2_A,pdockq2_B, … andpdockq2_lig(the last chain pdockq2) in the dataframe.LIS_pep(): compute the Local Interaction Score (LIS) between the receptor chain(s) and the ligand/peptide chain (last one). Add the columnsLIS_rec_pepandLIS_pep_recin the dataframe.
Example:
from af_analysis import docking
#extract_pae_pep
docking.pae_pep(my_data)
#compute_pdockq2_lig
docking.pdockq2_lig(my_data)
#compute_LIS_pep
docking.LIS_pep(my_data)
#extract_plddt_pep
docking.plddt_pep(my_data)
Plots
Interactive Visualization
At first approach the user can visualize the pLDDT, PAE matrix and the model scores.
The show_info() function displays the scores of the models, as well as the pLDDT
plot and PAE matrix in a interactive way.
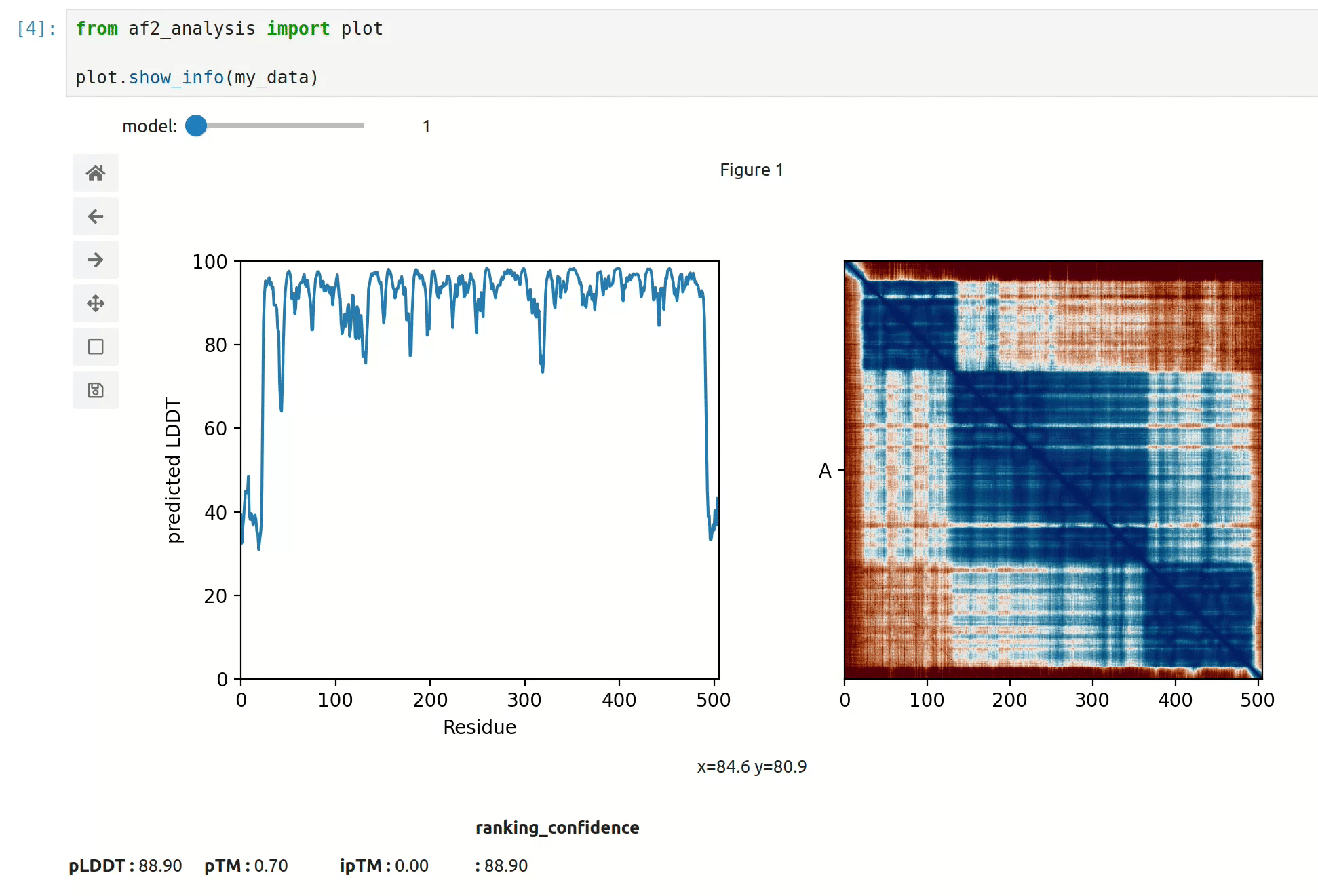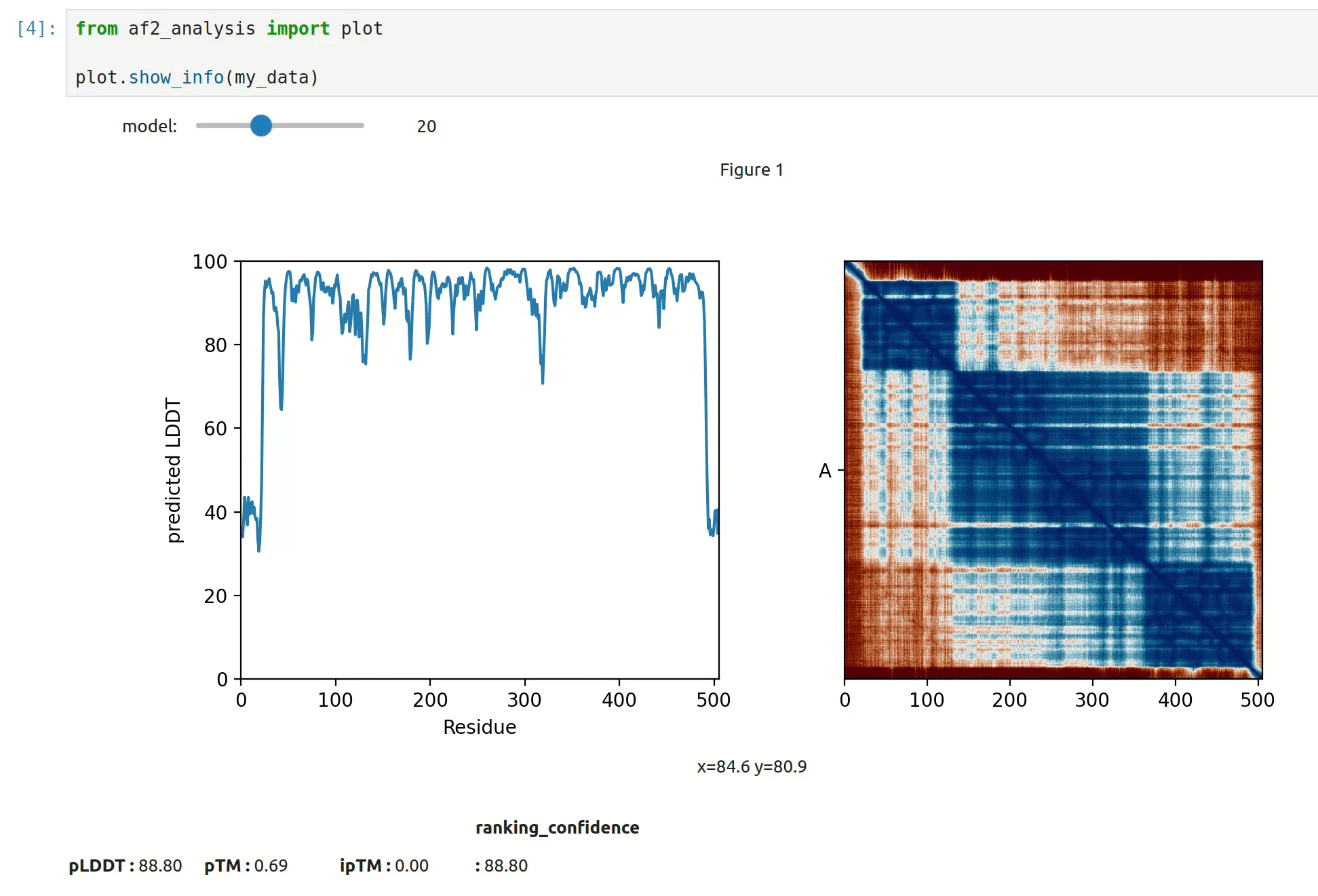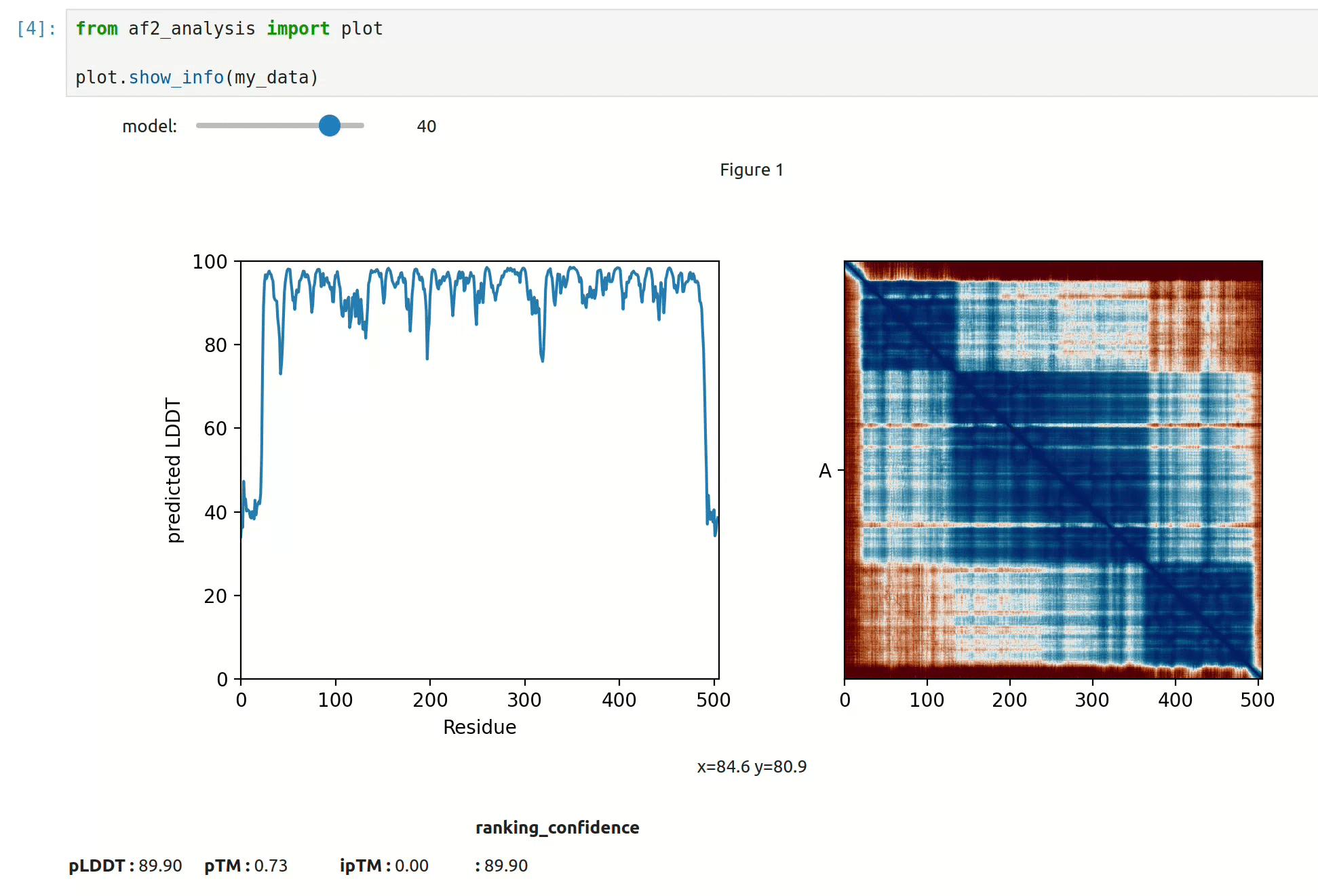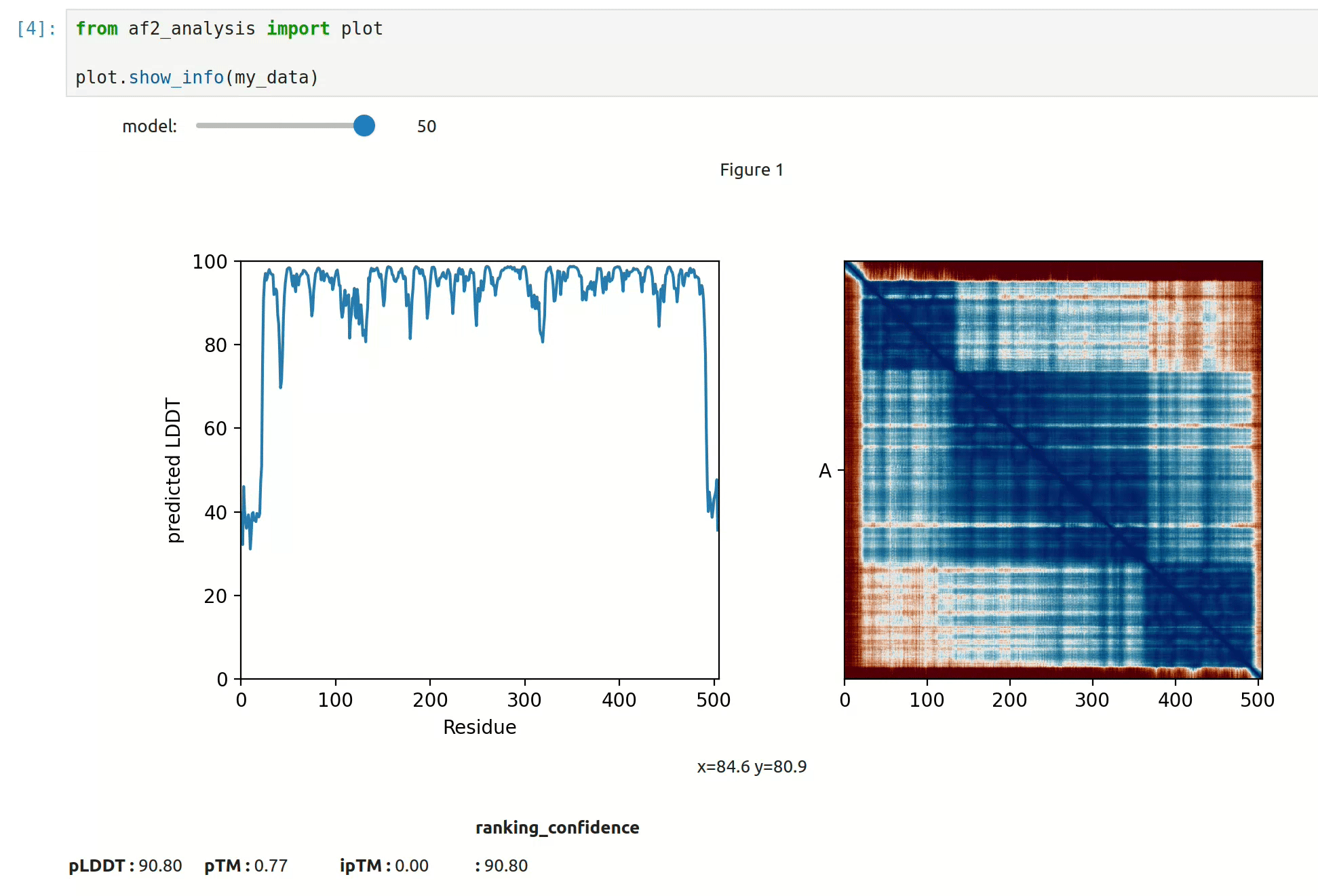
MSA Plot
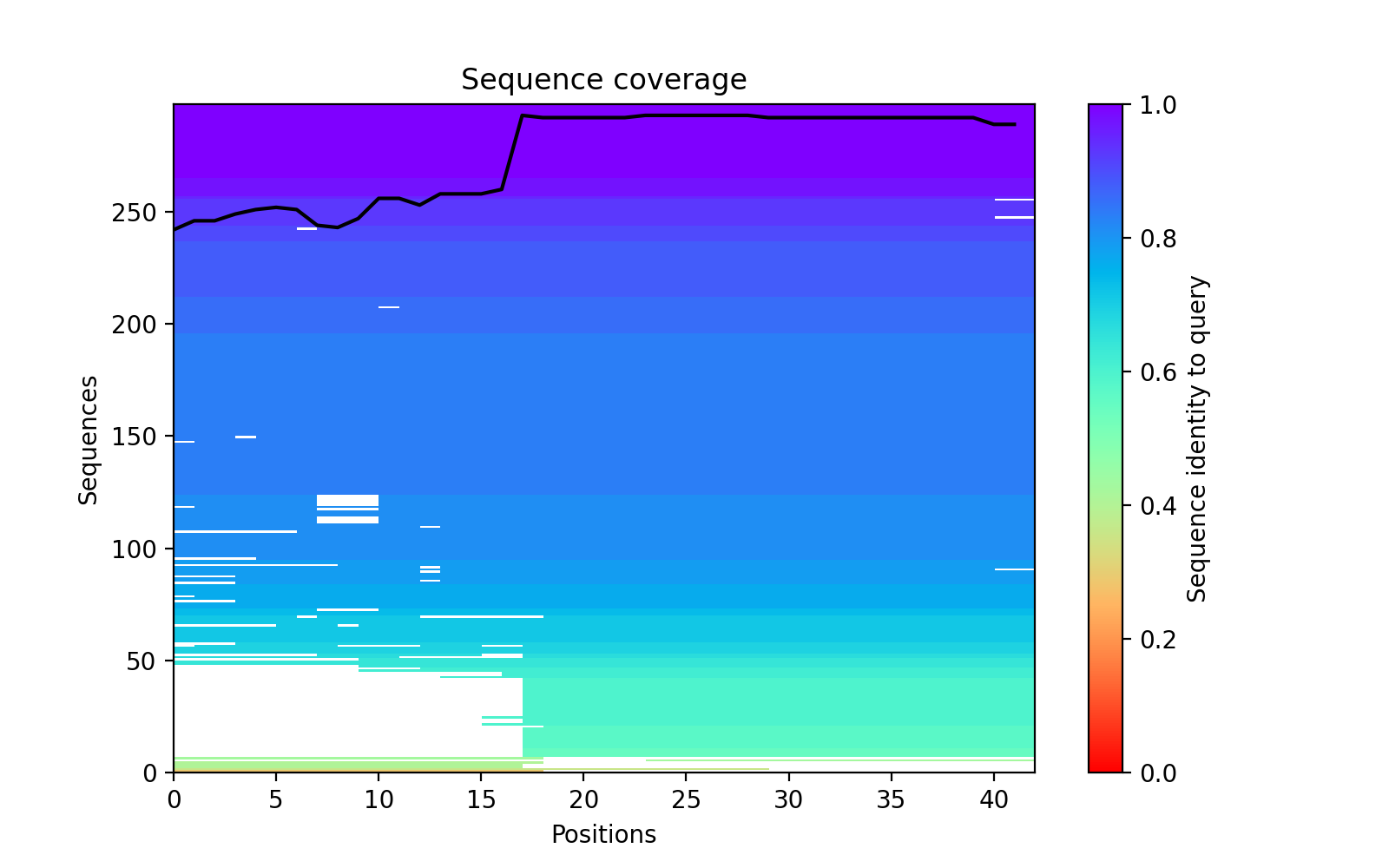
The plot_msa() function generates a multiple sequence alignment (MSA) plot for the
predicted models. The MSA plot shows the sequence conservation of the predicted models,
highlighting regions of high and low conservation.
my_data.plot_msa()
pLDDT Plot
The plot_plddt() function generates a pLDDT plot for the predicted models. The pLDDT
plot shows the per-residue local distance difference test (pLDDT) score for each residue
in the predicted models, highlighting regions of high and low model confidence.
you can plot all models plddt at once:
my_data.plot_plddt()
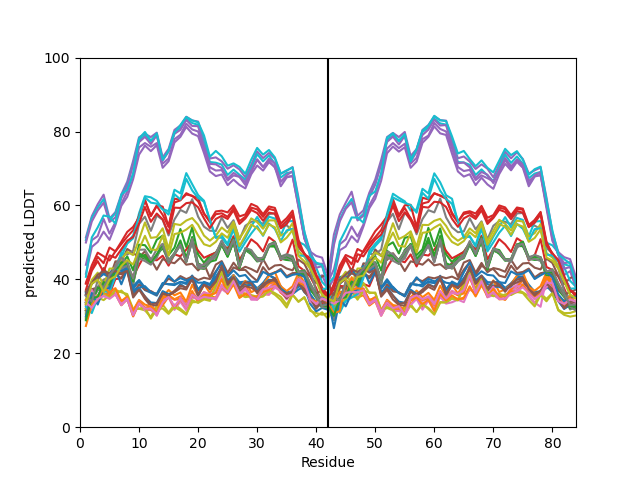
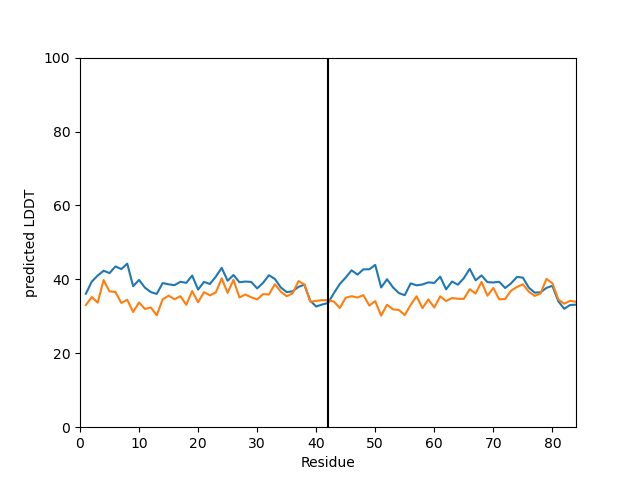
or you can plot specific models plddt:
my_data.plot_plddt([0,1])
PAE Plot
The plot_pae() function generates a predicted aligned error (PAE) plot for the
predicted models. The PAE plot shows the per-residue predicted aligned error for each
residue in the predicted models, highlighting regions of high and low model accuracy.
best_model_index = my_data.df['ranking_confidence'].idxmax()
my_data.plot_pae(best_model_index)
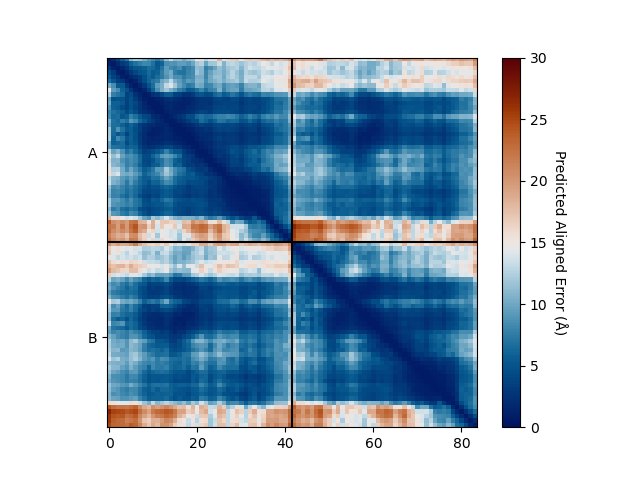
3D Structure Visualization
The show_3d() function displays the 3D structure of the predicted models using the nglview
package. The 3D structure visualization allows users to interactively explore the predicted models
and compare them with the experimental structure.
my_data.show_3d(my_data.df['ranking_confidence'].idxmax())
Clustering
This approach aims to address the challenge of managing and analyzing the large number of models (e.g., 10.000) produced for each protein complex, especially since these models often exhibit structural redundancies.
To do so, the user can use the clustering module to cluster the models
based on their structural similarity. The user can choose:
a selection to align model structures, e.g.
"backbone and chain A"a selection to calculate the RMSD matrices, e.g. ,
"backbone and chain B",a threshold value to determine the number of clusters.
RMSD can be scaled using Björn Wallner method:
with \(RMS\) the RMSD matrix and \(di\) a scaling factor of 8.5 Å.
From the distance matrix (scaled or not), an ascending hierarchical classification is computed to determine the clusters based on the distance threshold.
from af_analysis import clustering
clustering.hierarchical(my_data.df, threshold=2.5)
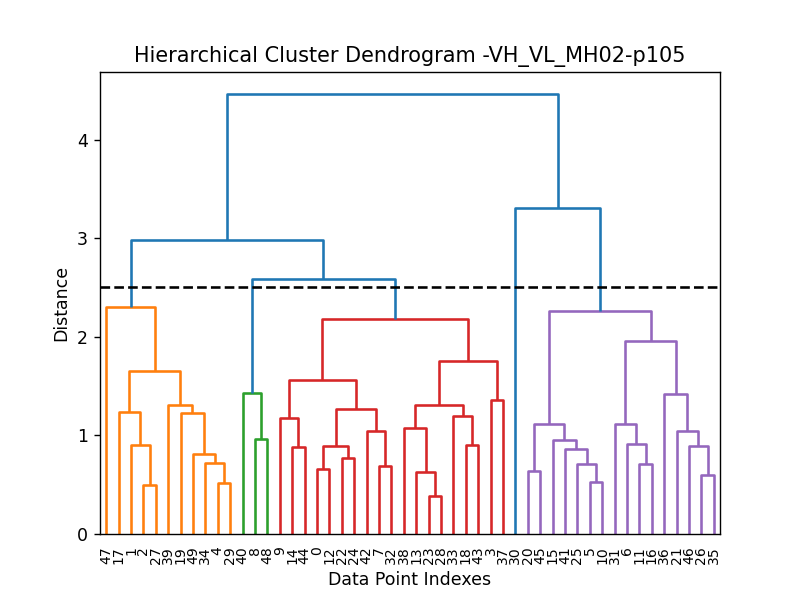
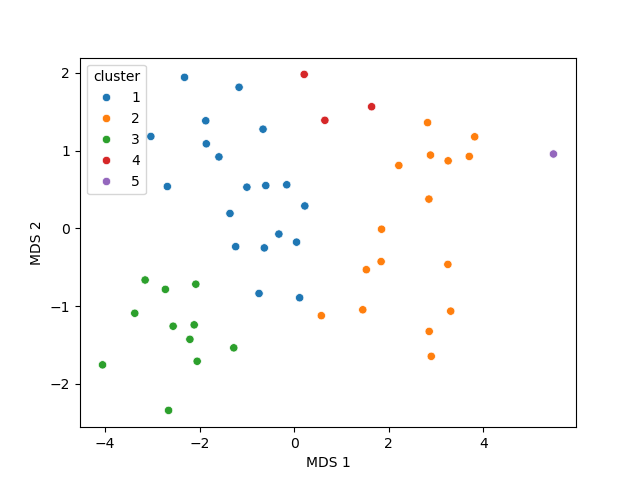
A multidimensional scaling (MDS) coordinates can be computed from the distance matrix
to visualize a 2D projection of the clusters, this coordinates are added in the dataframe
in column MDS 1 and MDS 2.
sns.scatterplot(data=my_data.df, x='MDS 1', y='MDS 2', hue='cluster')
Custom Analysis functions
The user can define custom analysis functions to compute additional metrics or visualizations.
In this example we use the pdb_cpp package to define the
contact_number() function which take a pdb file as input and
compute the number of contacts between chains.
# Here we use the `pdb_cpp` package to deal with coordinates file
import pdb_cpp
def contact_number(pdb, cutoff=8.0):
# Compute the number of contacts in the interface
coor = pdb_cpp.Coor(pdb)
chains = np.unique(coor.chain)
contact_num = 0
for chain in chains:
coor_interface = coor.select_atoms(
f"name CA and chain {chain} and within {cutoff} of not chain {chain}")
contact_num += coor_interface.len
return contact_num
The custom analysis function can then be applied easily to the dataframe:
# Apply the custom analysis function to the dataframe
contact_list = []
for pdb in my_data.df.pdb:
contact_list.append(contact_number(pdb))
# Add the contact number to the dataframe
my_data.df['contact_num'] = contact_list
The custom analysis results are then stored in the dataframe as the contact_num
column and can be used for further analysis.
FTDMP Scores
The af_analysis package provides a simple interface to extract docking score
computed using the FTDMP software.
First you need to install the ftdmp package:
git clone https://github.com/kliment-olechnovic/ftdmp.git
cd ./ftdmp
./core/build.bash
and then install dependencies using conda:
conda env create -f ftdmp_environment_for_conda.yml
Then you can use the FTDMP sofware to compute different scores from the pdb files:
conda activate ftdmp
ls MY_AF_DIRECTORY/*.pdb | ~/Documents/Code/ftdmp/ftdmp-qa-all --workdir ftdmp_beta_amyloid_dimer
you can then extract the scores using the analysis.extract_ftdmp() function in a
python script or a jupyter notebook :
import af_analysis
from af_analysis import analysis
my_data = af_analysis.Data('MY_AF_RESULTS_DIR')
# Extract the FTDMP scores
analysis.extract_ftdmp(my_data, ftdmp_path='ftdmp_beta_amyloid_dimer')
If you use the FTDMP software, please cite:
Olechnovič K, Banciul R, Dapkūnas J, Venclovas Č. (2025) FTDMP: A Framework for Protein-Protein, Protein-DNA, and Protein-RNA Docking and Scoring. Proteins. doi: 10.1002/prot.26792. PubMed PMID: 39748638.
Scoring of protein-protein interfaces using the VoroIF-jury algorithm and details of this algorithm are published in the CASP16 article:
Olechnovič K, Valančauskas L, Dapkūnas J, Venclovas Č. (2023) Prediction of protein assemblies by structure sampling followed by interface-focused scoring. Proteins; 91:1724–1733. doi: 10.1002/prot.26569. PubMed PMID: 37578163.
Citing this work
If you use the code of this package, please cite:
- Reguei A and Murail S. Af-analysis: a Python package for Alphafold analysis.Journal of Open Source Software (2025) doi: 10.21105/joss.07577
@Article{reguei_af-analysis_2025,
title = {Af-analysis: a {Python} package for {Alphafold} analysis},
volume = {10},
issn = {2475-9066},
shorttitle = {Af-analysis},
url = {https://joss.theoj.org/papers/10.21105/joss.07577},
doi = {10.21105/joss.07577},
language = {en},
number = {107},
urldate = {2025-03-14},
journal = {Journal of Open Source Software},
author = {Reguei, Alaa and Murail, Samuel},
month = mar,
year = {2025},
pages = {7577},
}